Introduction#
What is ambient signal?#
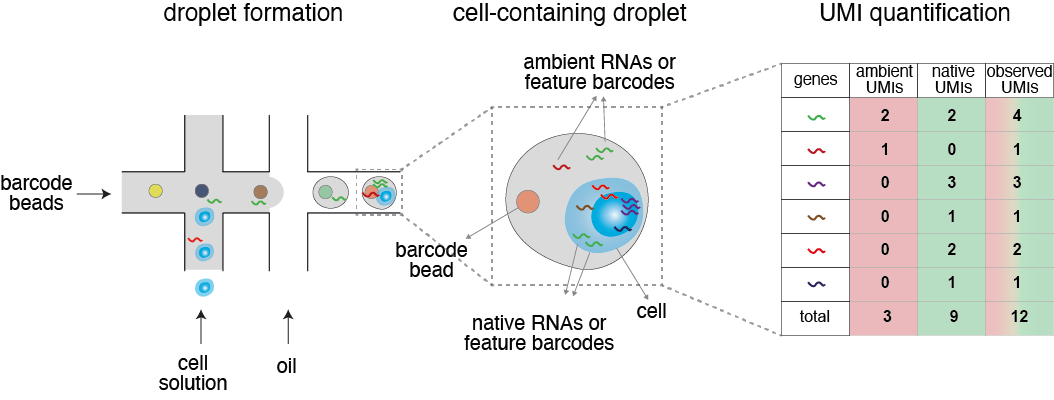
When preparing a single-cell solution, cell lysis releases RNA or protein counts that become encapsulated by droplets. These exogenous molecules are mixed with native ones and barcoded by the same 10x beads, leading to overestimated count data. The presence of this ambient signal can compromise downstream analysis and even introduce significant bias in certain cases, such as scCRISPR-seq and cell multiplexing.
The design of scAR#
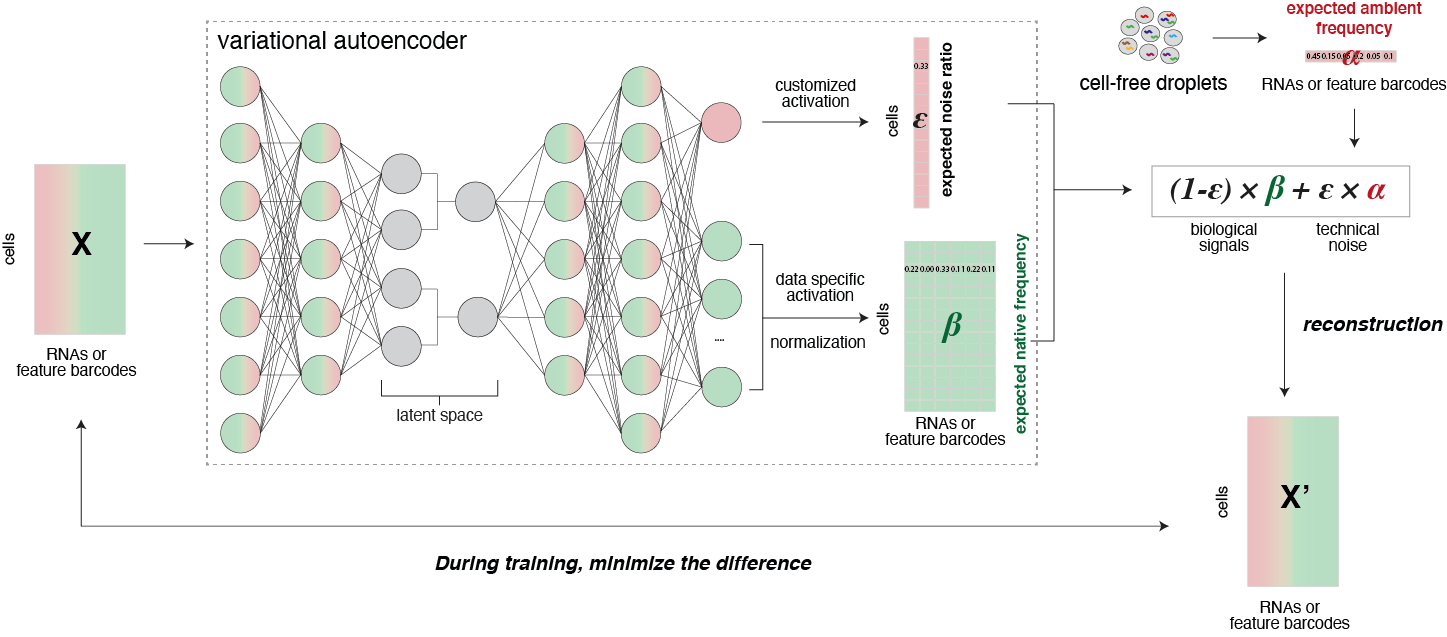
scAR employs a latent variable model that represents both biological and technical components in the observed count data. The model is developed under the ambient signal hypothesis, where the probability of each ambient transcript’s occurrence can be estimated empirically from cell-free droplets. The model has two hidden variables, namely the contamination level per cell and the probability of native transcript occurrence. With these three parameters, scAR can reconstruct noisy observations. We train neural networks, specifically the variational autoencoder, to learn the hidden variables by minimizing the differences between the reconstructions and the original noisy observations. Once the model converges, contamination levels and native expression are inferred, and downstream analysis can be performed using these values. For more information, please refer to our manuscript [Sheng2022].
What types of data that scAR can process?#
We validated the effectiveness of scAR across a range of droplet-based single-cell omics technologies, including scRNAseq, scCRISPR-seq, CITEseq, and scATACseq. scAR was able to remove ambient mRNA, assign sgRNAs, assign cell tags, clean noisy protein counts (ADT), and clean peak counts, resulting in significant improvements in data quality in all tested datasets. Notably, scAR was able to recover a substantial proportion (33% to 50%) of cells in scCRISPR-seq and cell multiplexing experiments. Given that ambient contamination is a common issue in droplet-based single-cell omics, particularly in complex experiments or samples, scAR represents a viable solution for addressing this challenge.
What are the alternative apporaches?#
Several methods exist for modeling noise in single-cell omics data. In general, these methods can be classified into two categories: those that deal with background noise and those that model stochastic noise. Some examples of these methods are provided below.
Background noise |
Stachastic noise |
||||
|---|---|---|---|---|---|
CellBender [Fleming2023] |
scVI [Lopez2018] |
||||
SoupX [Young2020] |
DCA [Eraslan2019] |
||||
DecontX [Yang2020] |
|||||
totalVI (protein counts) [Gayoso2021] |
|||||
DSB (protein counts) [Mulè2022] |