Tutorials#
There are two ways to run scar. For Python users, we recommend the Python API; for R users, we recommend the command line tool.
Run scar with Python API#
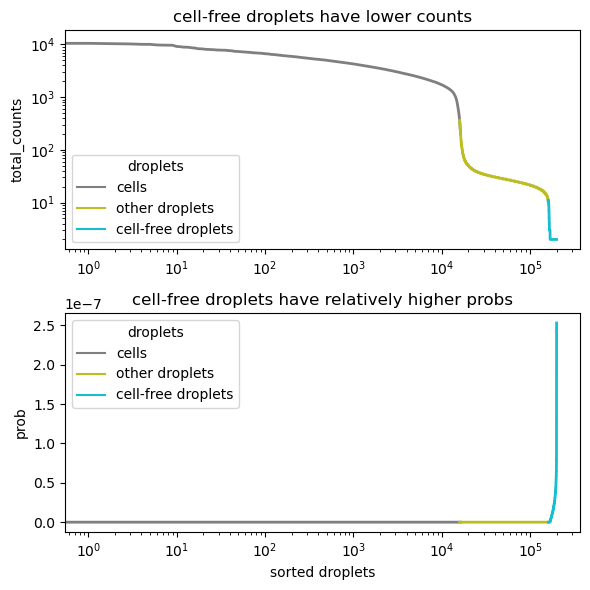
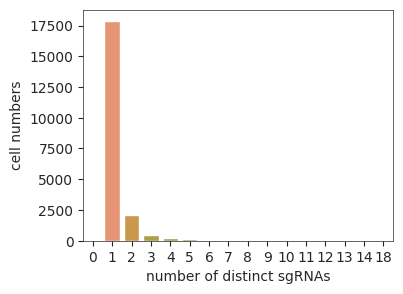
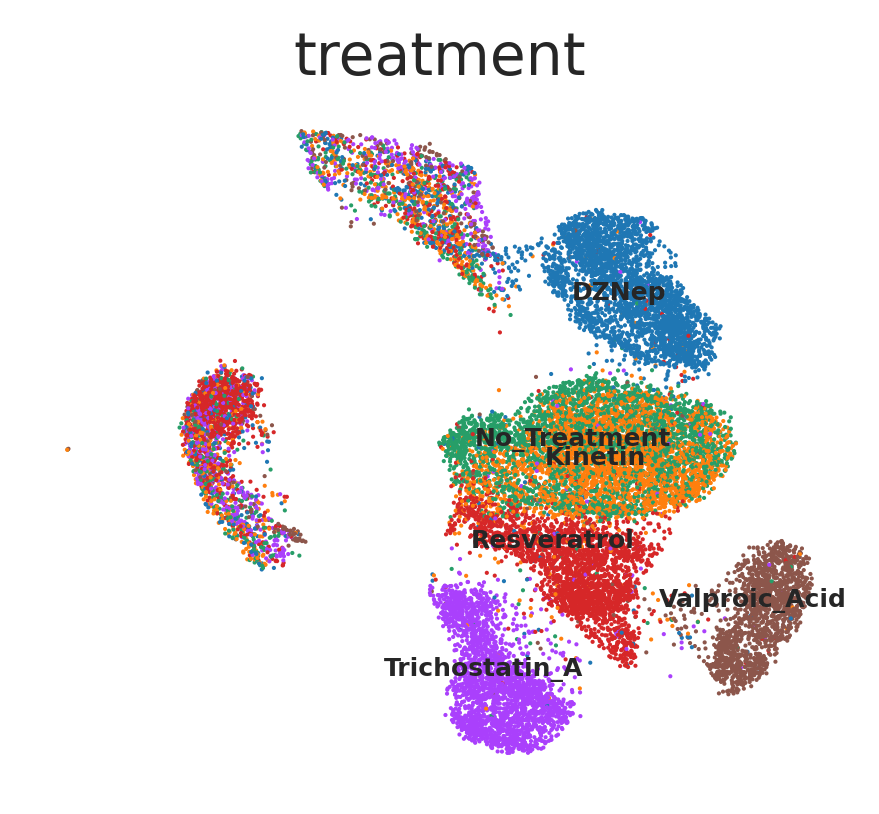
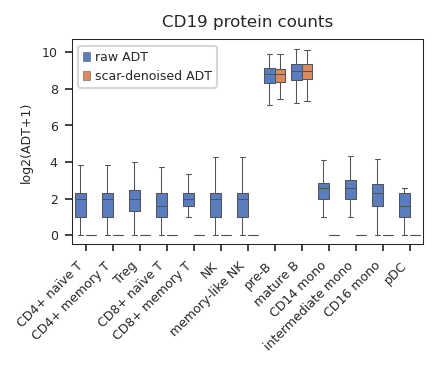
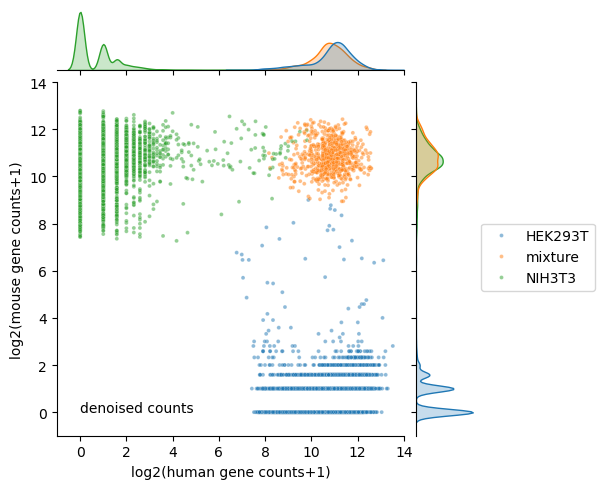
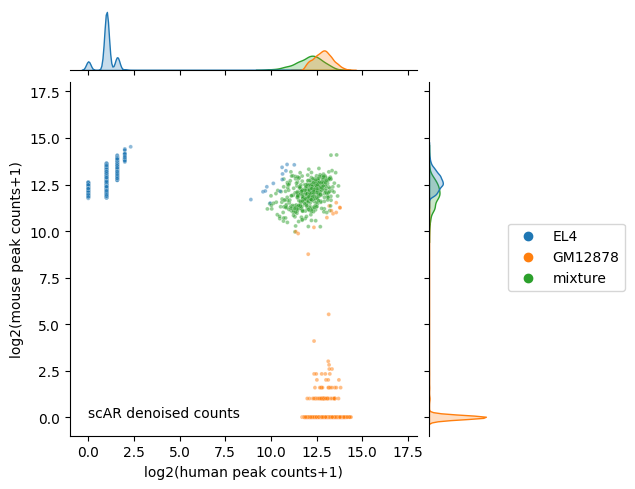

Run scar with the command line tool#
The command line tool supports two formats of input.
Use .h5 files as the input#
We can use the output of cellranger count filtered_feature_bc_matrix.h5 as the input for scar:
scar filtered_feature_bc_matrix.h5 -ft feature_type -o output
filtered_feature_bc_matrix.h5, a filtered .h5 file produced by cellranger count.
feature_type, a string, either ‘mRNA’ or ‘sgRNA’ or ‘ADT’ or ‘tag’ or ‘CMO’ or ‘ATAC’.
Note
The ambient profile is calculated by averaging the cell pool under this mode. If you want to use a more accurate ambient profile, please consider calculating it and using .pickle files as the input, as detailed below.
The output folder contains an h5ad file:
output
└── filtered_feature_bc_matrix_denoised_feature_type.h5ad
The h5ad file can be read by scanpy.read as an anndata object:
anndata.X, denosed counts.
anndata.obs[’
noise_ratio’], estimated noise ratio per cell.anndata.layers[’
native_frequencies’], estimated native frequencies.anndata.layers[’
BayesFactor’], bayesian factor of ambient contamination.anndata.obs[’
sgRNAs’ or ‘tags’], optional, feature assignment, e.g., sgRNA, tag, CMO, and etc..
Use .pickle files as the input#
We can also run scar by:
scar raw_count_matrix.pickle -ft feature_type -o output
raw_count_matrix.pickle, a file of raw count matrix (MxN) with cells in rows and features in columns.
cells |
gene_0 |
gene_1 |
… |
gene_y |
|---|---|---|---|---|
cell_0 |
12 |
3 |
… |
82 |
cell_1 |
13 |
0 |
… |
78 |
cell_2 |
35 |
30 |
… |
170 |
… |
… |
… |
… |
… |
cell_x |
16 |
5 |
… |
112 |
feature_type, a string, either ‘mRNA’ or ‘sgRNA’ or ‘ADT’ or ‘tag’ or ‘CMO’ or ‘ATAC’.
Note
An extra argument ambient_profile is recommended to achieve deeper noise reduction.
ambient_profile represents the probability of occurrence of each ambient transcript and can be empirically estimated by averging cell-free droplets.
genes |
ambient profile |
|---|---|
gene_0 |
.0003 |
gene_1 |
.00004 |
gene_2 |
.00003 |
… |
… |
gene_y |
.0012 |
Warning
ambient_profile should sum to one. The gene order should be consistent with raw_count_matrix.
For other optional arguments and parameters, run:
scar --help
The output folder contains four (or five) files:
output
├── denoised_counts.pickle
├── expected_noise_ratio.pickle
├── BayesFactor.pickle
├── expected_native_freq.pickle
└── assignment.pickle
In the folder structure above:
expected_noise_ratio.pickle, estimated noise ratio.denoised_counts.pickle, denoised count matrix.BayesFactor.pickle, bayesian factor of ambient contamination.expected_native_freq.pickle, estimated native frequencies.assignment.pickle, optional, feature assignment, e.g., sgRNA, tag, and etc..