Generate synthetic single-cell datasets#
Module to generate synthetic datasets with ambient contamination
Classes:
|
Generate synthetic single-cell RNAseq data with ambient contamination |
|
Generate synthetic ADT count data for CITE-seq with ambient contamination |
|
Generate synthetic sgRNA count data for scCRISPRseq with ambient contamination |
- class scar.main._data_generater.scrnaseq(n_cells, n_celltypes, n_features, n_total_molecules=8000, capture_rate=0.7)#
Generate synthetic single-cell RNAseq data with ambient contamination
- Parameters:
n_cells (int) – number of cells
n_celltypes (int) – number of cell types
n_features (int) – number of features (mRNA)
n_total_molecules (int, optional) – total molecules per cell, by default 8000
capture_rate (float, optional) – the probability of being captured by beads, by default 0.7
Examples
import numpy as np from scar import data_generator n_features = 1000 # 1000 genes, bad visualization with too big number n_cells = 6000 # cells n_total_molecules = 20000 # total mRNAs n_celltypes = 8 # cell types np.random.seed(8) scRNAseq = data_generator.scrnaseq(n_cells, n_celltypes, n_features, n_total_molecules=n_total_molecules) scRNAseq.generate(dirichlet_concentration_hyper=1) scRNAseq.heatmap(vmax=5)
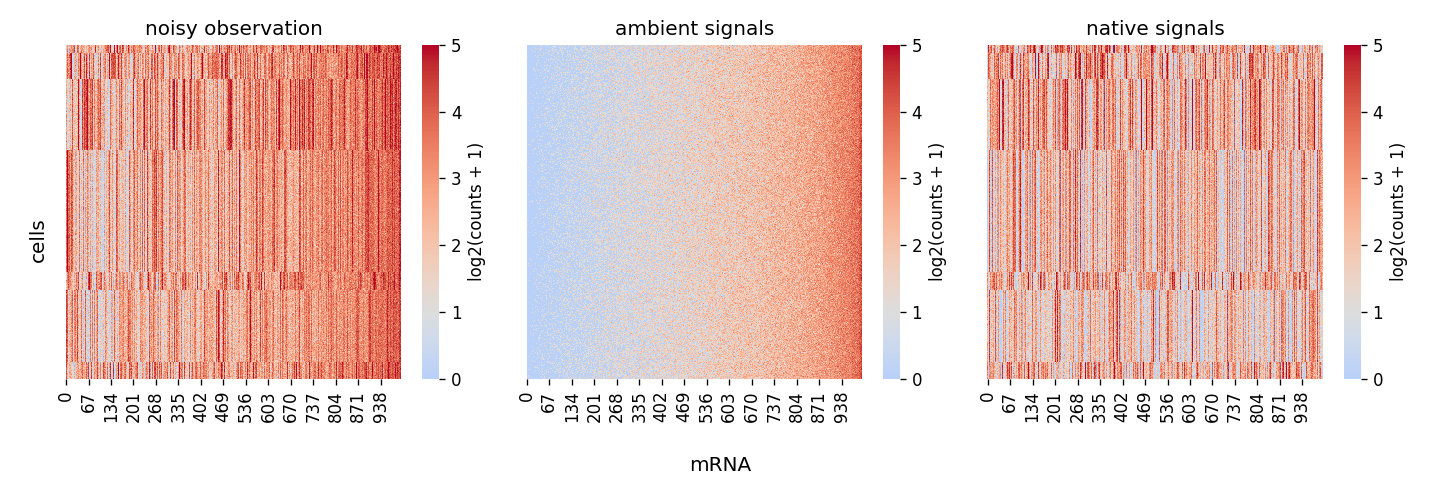
Attributes:
int, number of cells
int, number of cell types
int, number of features (mRNA, sgRNA, ADT, tag, CMO, and etc.)
int, number of total molecules per cell
float, the probability of being captured by beads
vector, observed counts
vector, the probability of occurrence of each ambient transcript
matrix, the onehot expression of the identity of cell types
vector, contamination level per cell
vector, the identity of cell types
matrix, the real ambient signals
matrix, the real native signals
matrix, the frequencies of the real native signals
vector, the total observed counts per cell
matrix, synthetic cell-free droplets
Methods:
generate([dirichlet_concentration_hyper])Generate a synthetic scRNAseq dataset.
heatmap([feature_type, return_obj, figsize, ...])Heatmap of synthetic data.
- n_cells#
int, number of cells
- n_celltypes#
int, number of cell types
- n_features#
int, number of features (mRNA, sgRNA, ADT, tag, CMO, and etc.)
- n_total_molecules#
int, number of total molecules per cell
- capture_rate#
float, the probability of being captured by beads
- obs_count#
vector, observed counts
- ambient_profile#
vector, the probability of occurrence of each ambient transcript
- cell_identity#
matrix, the onehot expression of the identity of cell types
- noise_ratio#
vector, contamination level per cell
- celltype#
vector, the identity of cell types
- ambient_signals#
matrix, the real ambient signals
- native_signals#
matrix, the real native signals
- native_profile#
matrix, the frequencies of the real native signals
- total_counts#
vector, the total observed counts per cell
- empty_droplets#
matrix, synthetic cell-free droplets
- generate(dirichlet_concentration_hyper=0.05)#
Generate a synthetic scRNAseq dataset.
- Parameters:
dirichlet_concentration_hyper (None or real, optional) – the concentration hyperparameters of dirichlet distribution. Determining the sparsity of native signals. If None, 1 / n_features, by default 0.005.
- Return type:
After running, several attributes are added
- heatmap(feature_type='mRNA', return_obj=False, figsize=(12, 4), vmin=0, vmax=10)#
Heatmap of synthetic data.
- Parameters:
feature_type (str, optional) – the feature types, by default “mRNA”
return_obj (bool, optional) – whether to output figure object, by default False
figsize (tuple, optional) – figure size, by default (15, 5)
vmin (int, optional) – colorbar minimum, by default 0
vmax (int, optional) – colorbar maximum, by default 10
- Returns:
if return_obj, return a fig object
- Return type:
fig object
- class scar.main._data_generater.citeseq(n_cells, n_celltypes, n_features, n_total_molecules=8000, capture_rate=0.7)#
Generate synthetic ADT count data for CITE-seq with ambient contamination
- Parameters:
n_cells (int) – number of cells
n_celltypes (int) – number of cell types
n_features (int) – number of distinct antibodies (ADTs)
n_total_molecules (int, optional) – number of total molecules, by default 8000
capture_rate (float, optional) – the probabilities of being captured by beads, by default 0.7
Examples
import numpy as np from scar import data_generator n_features = 50 # 50 ADTs n_cells = 6000 # 6000 cells n_celltypes = 6 # cell types # generate a synthetic ADT count dataset np.random.seed(8) citeseq = data_generator.citeseq(n_cells, n_celltypes, n_features) citeseq.generate() citeseq.heatmap()

Methods:
generate([dirichlet_concentration_hyper])Generate a synthetic ADT dataset.
heatmap([feature_type, return_obj, figsize, ...])Heatmap of synthetic data.
- generate(dirichlet_concentration_hyper=None)#
Generate a synthetic ADT dataset.
- Parameters:
dirichlet_concentration_hyper (None or real, optional) – the concentration hyperparameters of dirichlet distribution. If None, 1 / n_features, by default None
- Return type:
After running, several attributes are added
- heatmap(feature_type='ADT', return_obj=False, figsize=(12, 4), vmin=0, vmax=10)#
Heatmap of synthetic data.
- Parameters:
feature_type (str, optional) – the feature types, by default “ADT”
return_obj (bool, optional) – whether to output figure object, by default False
figsize (tuple, optional) – figure size, by default (15, 5)
vmin (int, optional) – colorbar minimum, by default 0
vmax (int, optional) – colorbar maximum, by default 10
- Returns:
if return_obj, return a fig object
- Return type:
fig object
- class scar.main._data_generater.cropseq(n_cells, n_celltypes, n_features)#
Generate synthetic sgRNA count data for scCRISPRseq with ambient contamination
- Parameters:
n_cells (int) – number of cells
n_celltypes (int) – number of cell types
n_features (int) – number of dinstinct sgRNAs
library_pattern (str, optional) –
the pattern of sgRNA libraries, three possibilities:
”uniform” - each sgRNA has equal frequency in the libraries”pyramid” - a few sgRNAs have significantly higher frequencies in the libraries”reverse_pyramid” - a few sgRNAs have significantly lower frequencies in the librariesBy default “pyramid”.noise_ratio (float, optional) – global contamination level, by default 0.005
average_counts_per_cell (int, optional) – average total sgRNA counts per cell, by default 2000
doublet_rate (int, optional) – doublet rate, by default 0
missing_rate (int, optional) – the fraction of droplets which have zero sgRNAs integrated, by default 0
Examples
import numpy as np from scar import data_generator n_features = 100 # 100 sgRNAs in the libraries n_cells = 6000 # 6000 cells n_celltypes = 1 # single cell line # generate a synthetic sgRNA count dataset np.random.seed(8) cropseq = data_generator.cropseq(n_cells, n_celltypes, n_features) cropseq.generate(noise_ratio=0.98) cropseq.heatmap(vmax=6)

Attributes:
vector, sgRNA frequencies in the libraries
Methods:
generate([dirichlet_concentration_hyper, ...])Generate a synthetic sgRNA count dataset.
heatmap([feature_type, return_obj, figsize, ...])Heatmap of synthetic data.
- sgrna_freq#
vector, sgRNA frequencies in the libraries
- generate(dirichlet_concentration_hyper=None, library_pattern='pyramid', noise_ratio=0.96, average_counts_per_cell=2000, doublet_rate=0, missing_rate=0)#
Generate a synthetic sgRNA count dataset.
- Parameters:
library_pattern (str, optional) – library pattern, by default “pyramid”
noise_ratio (float, optional) – global contamination level, by default 0.005
average_counts_per_cell (int, optional) – average total sgRNA counts per cell, by default 2000
doublet_rate (int, optional) – doublet rate, by default 0
missing_rate (int, optional) – the fraction of droplets which have zero sgRNAs integrated, by default 0
- Return type:
After running, several attributes are added
- heatmap(feature_type='sgRNAs', return_obj=False, figsize=(12, 4), vmin=0, vmax=7)#
Heatmap of synthetic data.
- Parameters:
feature_type (str, optional) – the feature types, by default “sgRNAs”
return_obj (bool, optional) – whether to output figure object, by default False
figsize (tuple, optional) – figure size, by default (15, 5)
vmin (int, optional) – colorbar minimum, by default 0
vmax (int, optional) – colorbar maximum, by default 10
- Returns:
if return_obj, return a fig object
- Return type:
fig object