Denoising ADT#
In this tutorial, we go through steps for ADT denoising in CITE-seq.
We will use the 10xPBMC5k CITE-seq dataset, which contained about five thousands cells stained with a panel of 32 antibodies. The original dataset was downloaded from 10x genomics dataset, and the processed and annotation data is available at scAR-reproducibility/data
Note
To run this notebook on your device, you need to install .
Alternatively, you can also run this notebook on Colab ![]()
[ ]:
# Run this cell to install scar in Colab
# Skip this cell if running on your own device
%pip install scanpy
%pip install git+https://github.com/Novartis/scAR.git
%pip install matplotlib==3.1.3 # Specify this matplotlib version to avoid errors
[1]:
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
import scanpy as sc
from scar import model
import warnings
warnings.simplefilter("ignore")
Load data#
Cells were annotated using well-established markers, see the scAR manuscript for details. For the tutoring purpose, the processed file (CITEseq_PBMCs_5k.h5ad) is provided.
[2]:
!git clone https://github.com/CaibinSh/scAR-reproducibility.git
PBMCs5k = sc.read('scAR-reproducibility/data/CITEseq_PBMCs_5k.h5ad')
fatal: destination path 'scAR-reproducibility' already exists and is not an empty directory.
visualization of cell types
[3]:
sc.settings.set_figure_params(dpi=150,figsize = (3, 3.1))
sc.pl.umap(PBMCs5k, size=10, color=["subtype"],color_map="viridis",legend_loc="on data", frameon=False, legend_fontsize=5)

Raw count matrix#
Get a dataframe of ADT raw count as the input of scar
[4]:
raw_ADT = PBMCs5k[:, PBMCs5k.var_names.str.endswith('_TotalSeqB')].to_df()
raw_ADT.columns = raw_ADT.columns.str.replace('_TotalSeqB','')
raw_ADT.head()
[4]:
| CD3 | CD4 | CD8a | CD11b | CD14 | CD15 | CD16 | CD19 | CD20 | CD25 | ... | CD197 | CD274 | CD278 | CD335 | PD-1 | HLA-DR | TIGIT | IgG1_control | IgG2a_control | IgG2b_control | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCGGGTAGTTCAAACC-1 | 876.0 | 968.0 | 8.0 | 10.0 | 8.0 | 9.0 | 1.0 | 3.0 | 10.0 | 3.0 | ... | 20.0 | 2.0 | 29.0 | 6.0 | 6.0 | 14.0 | 4.0 | 3.0 | 3.0 | 2.0 |
| CTTTCGGTCCGCAACG-1 | 516.0 | 664.0 | 4.0 | 4.0 | 4.0 | 6.0 | 0.0 | 3.0 | 1.0 | 2.0 | ... | 15.0 | 1.0 | 26.0 | 5.0 | 5.0 | 8.0 | 1.0 | 1.0 | 3.0 | 0.0 |
| TCTACCGGTGTAAACA-1 | 666.0 | 837.0 | 5.0 | 10.0 | 6.0 | 5.0 | 1.0 | 4.0 | 7.0 | 4.0 | ... | 30.0 | 1.0 | 36.0 | 2.0 | 5.0 | 9.0 | 1.0 | 3.0 | 3.0 | 2.0 |
| ATAGACCCATAATGCC-1 | 352.0 | 709.0 | 3.0 | 7.0 | 8.0 | 12.0 | 3.0 | 4.0 | 4.0 | 2.0 | ... | 13.0 | 2.0 | 100.0 | 6.0 | 12.0 | 9.0 | 4.0 | 2.0 | 2.0 | 2.0 |
| TCTATACAGGGTATAT-1 | 289.0 | 861.0 | 7.0 | 3.0 | 8.0 | 4.0 | 2.0 | 4.0 | 9.0 | 21.0 | ... | 10.0 | 3.0 | 31.0 | 3.0 | 3.0 | 11.0 | 1.0 | 4.0 | 4.0 | 4.0 |
5 rows × 32 columns
Ambient profile#
Get a dataframe of ADT ambient profile as the input of scar
[5]:
ambient_profile = PBMCs5k.uns['ambient_profile']
ambient_profile.head()
[5]:
| ambient_profile | |
|---|---|
| CD3 | 0.049876 |
| CD4 | 0.048823 |
| CD8a | 0.023453 |
| CD11b | 0.083170 |
| CD14 | 0.107178 |
IMPORTANT
For denoising CITEseq data, we do not recommend running scar with the default setting (i.e., estimating the ambient profile by averaging cells). Instead, we advise calculating the ambient profile by averaging cell-free droplets. Accurately calculating the ambient profile is crucial for precise denoising.
For more details, see an example of calculating ambient profile.
scAR allows for denoising a specific subset of genes and samples. To save computational time, it is advisable to use filtered raw counts (e.g., xxx_filtered_feature_bc_matrix.h5).
Training#
[6]:
ADT_scar = model(raw_count = raw_ADT,
ambient_profile = ambient_profile, # Providing ambient profile is recommended for CITEseq; in other modes, you can leave this argument as the default value -- None
feature_type = 'ADT', # "ADT" or "ADTs" for denoising protein counts in CITE-seq
count_model = 'binomial', # Depending on your data's sparsity, you can choose between 'binomial', 'possion', and 'zeroinflatedpossion'
)
ADT_scar.train(epochs=500,
batch_size=64,
verbose=True
)
# After training, we can infer the true protein signal
ADT_scar.inference() # by defaut, batch_size=None, set a batch_size if getting GPU memory issue
2024-08-10 21:06:51|INFO|model|cuda is detected and will be used.
2024-08-10 21:06:51|INFO|VAE|Running VAE using the following param set:
2024-08-10 21:06:51|INFO|VAE|...denoised count type: ADT
2024-08-10 21:06:51|INFO|VAE|...count model: binomial
2024-08-10 21:06:51|INFO|VAE|...num_input_feature: 32
2024-08-10 21:06:51|INFO|VAE|...NN_layer1: 150
2024-08-10 21:06:51|INFO|VAE|...NN_layer2: 100
2024-08-10 21:06:51|INFO|VAE|...latent_space: 15
2024-08-10 21:06:51|INFO|VAE|...dropout_prob: 0.00
2024-08-10 21:06:51|INFO|VAE|...expected data sparsity: 0.90
2024-08-10 21:06:52|INFO|model|kld_weight: 1.00e-05
2024-08-10 21:06:52|INFO|model|learning rate: 1.00e-03
2024-08-10 21:06:52|INFO|model|lr_step_size: 5
2024-08-10 21:06:52|INFO|model|lr_gamma: 0.97
Training: 100%|██████████| 500/500 [02:33<00:00, 3.25it/s, Loss=1.0748e+02]
The denoised counts are saved in ADT_scar.native_counts and can be used for downstream analysis.
[7]:
denoised_ADT = pd.DataFrame(ADT_scar.native_counts.toarray(), index=raw_ADT.index, columns=raw_ADT.columns)
denoised_ADT.head()
[7]:
| CD3 | CD4 | CD8a | CD11b | CD14 | CD15 | CD16 | CD19 | CD20 | CD25 | ... | CD197 | CD274 | CD278 | CD335 | PD-1 | HLA-DR | TIGIT | IgG1_control | IgG2a_control | IgG2b_control | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCGGGTAGTTCAAACC-1 | 865 | 949 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 0 | 26 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| CTTTCGGTCCGCAACG-1 | 494 | 658 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 0 | 25 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| TCTACCGGTGTAAACA-1 | 651 | 857 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 0 | 34 | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
| ATAGACCCATAATGCC-1 | 358 | 722 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | ... | 0 | 0 | 96 | 0 | 8 | 0 | 0 | 0 | 0 | 0 |
| TCTATACAGGGTATAT-1 | 278 | 861 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 26 | ... | 0 | 0 | 32 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
5 rows × 32 columns
Visualization#
Plot setting
[8]:
from matplotlib import pylab
params = {'legend.fontsize': 6,
'legend.title_fontsize': 8,
'figure.facecolor':"w",
'figure.figsize': (6, 4.5),
'axes.labelsize': 6,
'axes.titlesize':8,
'axes.linewidth': 0.5,
'xtick.labelsize':6,
'ytick.labelsize':6,
'axes.grid':False,}
pylab.rc('font',**{'family':'serif','serif':['Palatino'],'size':10})
pylab.rcParams.update(params);
sns.set_palette("muted");
sns.set_style("ticks");
sns.despine(offset=4, trim=True);
<Figure size 900x675 with 0 Axes>
Boxplot of ADT counts per cell type#
Prepare a dataframe for visualization
[13]:
denoised_ADT_stacked = np.log2(denoised_ADT+1).stack().to_frame('log2(ADT+1)').rename_axis(['cells', 'ADTs']).reset_index().set_index('cells')
denoised_ADT_stacked = denoised_ADT_stacked.join(PBMCs5k.obs)
denoised_ADT_stacked['count'] = 'scar-denoised ADT'
raw_ADT_stacked = np.log2(raw_ADT+1).stack().to_frame('log2(ADT+1)').rename_axis(['cells', 'ADTs']).reset_index().set_index('cells') # log scale for visualization,
raw_ADT_stacked = raw_ADT_stacked.join(PBMCs5k.obs) # Add cell type information
raw_ADT_stacked['count'] = 'raw ADT'
ADTs_stacked = pd.concat([raw_ADT_stacked, denoised_ADT_stacked])
ADTs_stacked.head()
[13]:
| ADTs | log2(ADT+1) | celltype | subtype | count | |
|---|---|---|---|---|---|
| cells | |||||
| CCGGGTAGTTCAAACC-1 | CD3 | 9.776433 | T cells | CD4+ naïve T | raw ADT |
| CCGGGTAGTTCAAACC-1 | CD4 | 9.920353 | T cells | CD4+ naïve T | raw ADT |
| CCGGGTAGTTCAAACC-1 | CD8a | 3.169925 | T cells | CD4+ naïve T | raw ADT |
| CCGGGTAGTTCAAACC-1 | CD11b | 3.459432 | T cells | CD4+ naïve T | raw ADT |
| CCGGGTAGTTCAAACC-1 | CD14 | 3.169925 | T cells | CD4+ naïve T | raw ADT |
Boxplot visualization of ADT counts per cell type before and after denoising.
[14]:
marker = "CD19"
tmp = ADTs_stacked[ADTs_stacked['ADTs']==marker]
plt.figure(figsize=(3, 1.8))
g = sns.boxplot(x="subtype",
y="log2(ADT+1)",
data=tmp,
hue="count",
hue_order=['raw ADT', 'scar-denoised ADT'],
orient="v",
dodge=True,
width=0.8,
showfliers=False,
linewidth=0.5,
);
g.legend_.set_title('')
g.set(xlabel='',title=f'{marker} protein counts');
g.set_xticklabels(g.get_xticklabels(),rotation=45, ha='right');
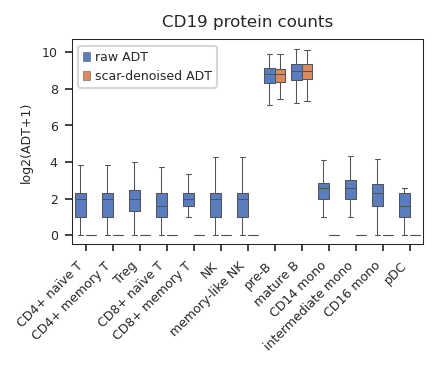
CD19 is a B cell specific marker, raw CD19 counts are detected in every cell type, while denoised counts are only observed in B cells.
Similarly, we observed ambient ADTs of other markers and scar can remove these ambient signals but preserve ‘true’ ADTs.
[15]:
for marker in ["CD3", "CD4", "CD8a", "CD11b", "PD-1", "IgG2a_control"]:
tmp = ADTs_stacked[ADTs_stacked['ADTs']==marker]
plt.figure(figsize=(3, 1.8))
g = sns.boxplot(x="subtype",
y="log2(ADT+1)",
data=tmp,
hue="count",
hue_order=['raw ADT', 'scar-denoised ADT'],
orient="v",
dodge=True,
width=0.8,
showfliers=False,
linewidth=0.5,
);
g.legend_.set_title('')
g.set(xlabel='', title=f'{marker} protein counts');
g.set_xticklabels(g.get_xticklabels(),rotation=45, ha='right')






UMAP of ADT and RNA#
match ADT name and transcript name
[16]:
prot2gene = PBMCs5k.uns['prot2gene']
Expression of mRNA
[17]:
mRNA = PBMCs5k[:, list(prot2gene.values())].to_df()
mRNA.columns = mRNA.columns.astype(str)+'_RNA'
[18]:
raw_ADT.columns = 'raw_' + raw_ADT.columns.astype(str)
denoised_ADT.columns = 'denoised_' + denoised_ADT.columns.astype(str)
PBMCs5k.obs = PBMCs5k.obs.join(mRNA).join(raw_ADT).join(denoised_ADT)
[19]:
sc.settings.set_figure_params(dpi = 150, figsize = (1.5, 1.5), fontsize = 6)
for marker in ["CD3", "CD4", "CD8a", "CD19", "CD11b", "PD-1"]:
raw_prot_label = f'raw_{marker}'
denoised_prot_label = f'denoised_{marker}'
RNA_label = f'{prot2gene[marker]}_RNA'
sc.pl.umap(PBMCs5k,
size = 5,
color = [raw_prot_label, denoised_prot_label, RNA_label],
color_map = "viridis",
frameon = False,
vmax = [8, 8, 3],
vmin = 0,
wspace = 0.1
)





